An estimated 100,000+ people in the US, the majority of whom are black or Hispanic, suffer from a hereditary condition, which causes changes in the structure and function of red blood cells. Sickle Cell Disease (SCD), which encompasses a group of conditions, Sickle Cell Anemia (SCA) being only one of them, is both life-threatening and painful; the condition reduces the life-span of those who have inherited it by decades.
While suffering from chronic SCD is bad enough, those afflicted sometimes face very serious bouts of acute flare-ups called sickle-cell crises, which are known to be extraordinarily painful. Powerful pain medications are essential during a crisis. In a rational world, one would assume that patients with SCD who are in a sickle-cell crisis would be given whatever drugs are necessary to cope with the pain, especially since are victims of a well-established condition, not addicts faking pain simply to get the drugs to which they are addicted. Yet, even this is false.
Today's war on pain patients doesn't actively discriminate; it provides equal opportunity inhumanity to all. Or does it? While I don't believe for a second that current policies of denying adequate medical care to pain patients were put in place to single out specific groups or race, for Blacks with SCD the end result is the same - "unintended racism." All pain patients suffer, but some suffer more. Perhaps no one knows this better than Adrienne Kincaid.
Ms. Kincaid, a resident of Euclid, Ohio, is the founder of Kincaid's Kindred Spirits, Inc., a support group for Sickle Cell anemia sufferers which she formed in Cleveland in 2004. Ms. Kincaid has kindly agreed to speak with me about the extra burden that is placed on the backs of sufferers of SCD. Ms. Glinda Dames-Fincher [GDF], a medical technician in the group who also suffers from SCD, also contributed to this interview.
What you are about to read is very disturbing.
JB: Ms. Kincaid and Ms. Dames-Fincher, thank you very much for taking the time to speak with me. I have never written about racial or ethnic aspects of the "war on pain patients," but it would seem that blacks, who bear the brunt of SCD, are disproportionally being impacted; albeit it unintentionally by this war. Do you agree?
AK: Yes, I wholeheartedly agree. It has been shown in multiple studies that in the United States African Americans and Hispanics wait longer for pain medication, and are given less pain medication than Caucasians for the same illnesses and injuries. Other studies have shown that African Americans underreport pain, are more concerned with addiction, and less likely to abuse opioid drugs more frequently than whites. Yet, this message is not getting out.
Most research suggests that healthcare providers incorrectly believe that African Americans and Hispanics are more likely to abuse drugs than whites and so should have less access to these medications. (1,2,3,4) [GDF]
JB: Can you tell us about your support group and how you came to form it?
AK: One day in 2002 I was working, being a productive citizen and not relying on Social Security to support me, I received a shocking performance review. I was told that my illness was a hindrance to the development of the department. This made me furious and I began to wonder that if I felt this ill, but was still well enough to work for seven years, how someone who is even sicker and unable to work might feel.
So, that night I decided that I was going to start a support group for adults who suffer from SCD. It took two years to get all the pieces in place, but in July of 2004, we had our first support group meeting at the Euclid Library in Euclid, Ohio, and we haven't stopped moving since. There is much to do. In addition to supplying information about SCD we sponsored a bowling fundraiser, a cookout and our "Red & White Cell-abration" which is held in September - Sickle Cell Awareness Month. It turned out that an advocacy/support group was needed more than we thought.
JB: Why is that?
Because of funding cuts in SCD programs, and later, restrictions on pain medications, we as advocates had to become even more aggressive in bringing sickle cell awareness to the forefront. This requires more than just the victims speaking up. Outside help is also needed to help convey the plight of SCD sufferers, some of whom are afraid to speak up, often because of their own health issues. So, It is vitally important to get the family members educated and involved because SCD sufferers sometimes just don't have enough energy to keep going. But we can’t stop. It is our obligation to ensure that the next generation doesn't have to go through what we are now.
JB: It is no secret that pain patients, in general, are having more difficulty getting the medications they need. Does the same hold true for SCD sufferers?
AK: Yes it does! Pain meds for SCD sufferers are being cut in half by physicians because of fear of concerns about addiction and toxicity from the long-term use of opioids. Although research is ongoing to develop better pain drugs, these are far away. Meanwhile, we are left to suffer. And we certainly do.
JB: Can you describe a "typical" member of your group, for example, age, severity of disease, or degree of disability?
AK: Most of our group ranges from in age from between 20 and 60 years. The majority of the group has the most common form of sickle cell disease which is called SS Hemoglobin disease, but some have Sickle-Beta Thalassemia (5) and other forms of the disease such as Sickle-C disease (a milder form). As far as the severity of the disease, I don’t like to say this one is worse than that other; it is all bad and severely painful. You can have two people with the same type, but it may affect them differently. (5)
Sickie cell disease. Photo: Facebook.
JB: Since SCD has a wide range of severity can you compare what life is like for someone with "milder" disease with someone with a more severe form of the disease?
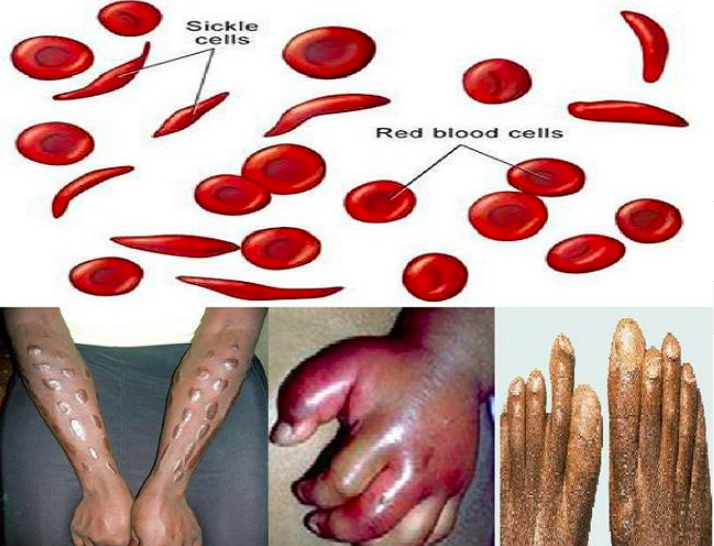
AK: SCD is a disease of the red blood cells. Normal red blood cells flow through the blood vessels and carry oxygen from the lungs to all parts of the body. But sickle cells do not carry oxygen well and they also change shape from round and flexible, to broken and sticky. The change in shape causes the defective cells to clog blood vessels and block oxygen supply, causing tissues and organ damage. This clogging is what causes the severe pain, tissue damage, internal organ failure, and death. People who have the milder form will not have as many hospital stays or crises.
JB: Can you give us an idea of what a person with a more severe disease will experience?
AK: It's awful. Some of the most severe symptoms are severe fatigue, strokes, kidney, heart, lung, liver or spleen failure, bone and joint damage, blindness, chronic leg wounds causing amputations, priapism (a painful sustained erection), neuropathy, life-threatening cases of pneumonia, and a weakened immune system which permits otherwise benign germs to cause infection. Although some of us end up in the hospital once or twice each month, most of us suffer the pain at home.
JB: How common is sickle-cell crisis? Can you give us an idea of what it is like?
AK: Sickle-Cell Crisis is very common. It has been described as being stabbed in the same place over and over and over. Or if you stub your toe on the corner of the table leg and just keep hitting it every time you walk by. It has also been described as having your bones crushed in a vice for hours and days at a time. [GDF]
JB: An increasingly common complaint of pain patients is that they feel like they are treated are treated like addicts looking for a fix. Have you or others in your group found this to be true? ? How are you and your group treated by physicians and emergency departments?
AK: This is so true and so sad. If we have adequate pain meds at home, we avoid the hospital whenever possible. But sometimes home pain control is inadequate and our only choice is the hospital. Even so, most of us wait at least two to three days before going because we know how we will be treated once we get there.
JB: Do you know people who suffer from a sickle-cell crisis and have been denied proper care?
AK: Of course. It happens all the time. We wait so long to go to the hospital when we are in a crisis because of the very poor treatment that we know we will receive. A wait of 12-plus hours in the emergency room is not uncommon. People have died from multiple organ failures, or "chest syndrome," - an acute heart and lung failure. Patients in the waiting room are often told that they are "drug seeking", and treated as addicts. Part of this is due to ignorance about SCD by the hospital staff and some is due to the demonization of pain patients in general.
JB: In your opinion, how did our health care system fail these patients?
AK: Some physicians have admitted that racism and classism in our healthcare system have caused sickle cell disease to be one of the most discriminated against diseases in the US. Sickle cell disease is found in all races and is the most common inherited blood disorder in the US and in the world. Other diseases that affect less than one-third as many people as SCD get three to 10 times more funding from the federal government and private sources for treatment and research. Cancer patients are treated by the same specialists (hematologist/oncologist) and in the same facilities as sickle cell patients. But cancer patients get much more compassionate and ethical care by doctors and nurses than do SCD patients. SCD is a lifelong, extremely painful and debilitating disease and shortens survival. Presently, the average age of death is in the late 40's to late 50's years of age, with many still dying as children and young adults. [GDF]
JB: What would you do to improve the management of SCD?
AK: First, the CDC and FDA must treat Sickle Cell Disease like cancer and lift restrictions on the dose of pain medication. Second, and possibly most important, the government and medical professional inspection and certification agencies, such as the Joint Commission on Accreditation of Healthcare Organizations, AHA, and CMS-Medicare & Medicaid must begin to enforce the use of the Sickle Cell Disease Treatment Guidelines that was published the NIH in 1984, and most recently updated in 2014.
Although, SCD has been taught in medical schools for more than 50 years and in nursing and other medical professions, too many patients encounter American licensed physicians and nurses who claim ignorance of basic knowledge of the disease and basic ER care. It is also essential that Congress to fully fund the 40 SCD treatment centers it voted for in 2004. They funded only 10, and since then we've lost funding for three of those. This is unacceptable.[GDF]
JB: Do you have a message for the bureaucrats, legislators, and law enforcement agencies?
AK: Yes. As previously stated, FDA and CDC must view SCD like cancer with regard to narcotic prescribing. Congress must equitably fund SCD treatment and research. It took almost four years to get the SCD treatment reauthorization passed through the House this year, and the amount of funding is still woefully inadequate.
It costs about $1 billion per year to treat SCD in America. If you seriously want to decrease that cost, then do what has been proven repeatedly for over 20 years to decrease ER visits and hospitalizations, and to improve quality of life and ability of the SCD affected to be employed. Provide care in specialized SCD treatment centers. Congress, please fully fund 40 SCD treatment centers as you voted for in 2004.
DEA, please stop targeting and bullying physicians who have given excellent, ethical, and appropriate care to pain patients for decades, and who now cannot treat any of them. It is causing the deaths of those who are not addicts and not criminals, but who are simply unfortunate enough to have a severely painful disease. The suicide rate in this population of patients is climbing. Please stop having knee-jerk reactions to America's problems, because it just makes the problems worse. Include all stakeholders in your decisions, and not just the privileged few (7).
NOTES:
(1) "Racial Disparities in Pain Management of Children With Appendicitis in Emergency Departments" JAMA Pediatrics, 2015 Nov;169(11):996-1002.
(2) "Pain and Ethnicity" AMA Journal of Ethics, May 2013, Volume 15, Number 5: 449-454;
(3) "The Opioid Drug Epidemic and Sickle Cell Disease: Guilt by Association" Pain Medicine 2016; 17: 1793–1798.
(4) American Society of Hematology - Current Issues in Sickle Cell Pain and Its Management, by Samir K. Ballas, 2007.
(5) Sickle-Beta Thalassemia is characterized by abnormally low levels of hemoglobin, the protein that carries oxygen throughout the body.
(6) Everyone has two hemoglobin genes, one inherited from each parent. Normally, you inherit a normal aka "A" (for normal adult) gene from each parent. If both parents each have one normal gene and one sickle gene, then they are "carriers" of sickle cell, but do not have the disease. In order to be afflicted by sickle cell disease, one must inherit one sickle hemoglobin gene from each parent.[GDF]
(7) See "Pain Advocacy Week."

Josh Bloom
Director of Chemical and Pharmaceutical Science
Dr. Josh Bloom, the Director of Chemical and Pharmaceutical Science, comes from the world of drug discovery, where he did research for more than 20 years. He holds a Ph.D. in chemistry.


